The Schuele lab is interested in the understanding of genetic causes and risk factors for neurodegeneration and the pathophysiological consequences leading to the clinical expression of Parkinson’s disease and related disorders.
Our projects range from clinical genetic family studies and human stem cell modeling of neuronal cell types or neurocircuits to translational approaches to ultimately treat Parkinson’s disease.
The Schuele lab has extensive expertise and a long history of human induced pluripotent stem cell modeling and gene discovery and has been a long-standing collaborator in several MJFF-funded LRRK2 and alpha-synuclein consortia.
Current work relates to genetic modifiers of LRRK2, gene dosage levels of alpha-synuclein, and ATXN-10 as a rare form of Parkinson’s disease/spinocerebellar ataxia.
Our projects range from clinical genetic family studies and human stem cell modeling of neuronal cell types or neurocircuits to translational approaches to ultimately treat Parkinson’s disease.
The Schuele lab has extensive expertise and a long history of human induced pluripotent stem cell modeling and gene discovery and has been a long-standing collaborator in several MJFF-funded LRRK2 and alpha-synuclein consortia.
Current work relates to genetic modifiers of LRRK2, gene dosage levels of alpha-synuclein, and ATXN-10 as a rare form of Parkinson’s disease/spinocerebellar ataxia.
Parkinson’s disease 'in-a-dish' and 'on-a-chip'
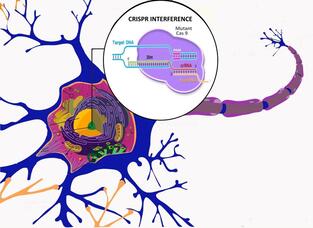 CRISPR interference in the neuron (Picture by Shivani Seshan)
CRISPR interference in the neuron (Picture by Shivani Seshan)
Skin-derived human stem cells build the foundation for in vitro modeling. Patient-derived stem cells can be differentiated into neurons, specifically neurons that produce and release dopamine resembling the neurons that die in the brain of Parkinson’s patients. This unique human cellular model allows replicating conditions as in the human brain.
In a collaborative project with VIB/KU Leuven and imec, we develop a novel neuronal microchip ('brain on a chip') to create a functional cortico-striato-nigral circuit to understand how these more complex systems can distinguish Parkinson’s disease from healthy controls. Check it out!
“Brain on a chip is a transformative, multidisciplinary and international collaboration
to approach next generation experimental research for Parkinson’s disease. This project
will give us deeper insight into functional network changes of the brain, that will allow us
to develop new therapies.” – Dr. J. William Langston
Clinical Professor, Dept. of Neurology & Neurological Sciences, Stanford University
In a collaborative project with VIB/KU Leuven and imec, we develop a novel neuronal microchip ('brain on a chip') to create a functional cortico-striato-nigral circuit to understand how these more complex systems can distinguish Parkinson’s disease from healthy controls. Check it out!
“Brain on a chip is a transformative, multidisciplinary and international collaboration
to approach next generation experimental research for Parkinson’s disease. This project
will give us deeper insight into functional network changes of the brain, that will allow us
to develop new therapies.” – Dr. J. William Langston
Clinical Professor, Dept. of Neurology & Neurological Sciences, Stanford University
Developing human iPSC resources for the scientific community
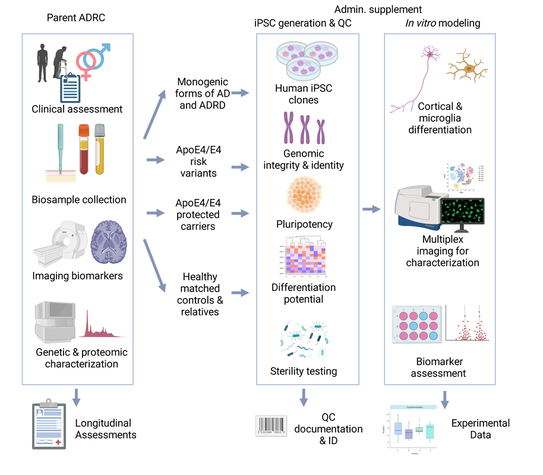
Roughly 10,000 human iPSC lines were created from 2008 to 2016, spanning diverse clinical and genetic conditions. Over the past decade, in vitro modeling utilizing iPSCs has displayed substantial potential.
However, variability and reproducibility is a critical challenge in human iPSC research. Biological factors, such as individual differences, genetic background, and cellular heterogeneity, contribute to this variability. Technical aspects, including variations in reprogramming efficiency, differentiation protocols, and culture conditions lead to reproducibility issues. This variability poses a substantial challenge in the accurate interpretation of data generated from iPSC models.
To overcome these barriers, we implement: 1. derivation of high-quality iPSC lines from consistent starting material with uniform non-integrating reprogramming techniques, 2. establishment of a high-quality protocols for iPSC differentiation along with clear guidelines for maintenance and storage, 3. define experimental endpoints that represent the benchmarks that a differentiating human iPSC must achieve to qualify as a differentiated neuron and 4. define robust indicators of the desired cell type, including disease biomarkers.
Since 2009, the Schuele lab has established a repository of human skin cells and fibroblasts derived from over 100 patients and healthy controls with genetic forms of Parkinson’s disease and related neurodegenerative diseases. This invaluable collection is currently housed within the Schuele lab and owned by the Michael J. Fox Foundation committed to promoting research accessibility and facilitating the broad dissemination of these invaluable cell lines across the scientific community.
The Schuele lab is affiliated with the Stanford Alzheimer Research Center (ADRC) and is supported by NIA to derive a human iPSC bank for the Stanford ADRC cohort from patients with monogenic forms of Alzheimer disease, ApoE4 genotypes with and without Alzheimer disease to study resistance and resilience, and matched healthy controls.
All human iPSCs are shared with the NINDS Human Cell and Data Repository (NHCDR), American Type Culture Collection (ATCC) or National Cell Repository for Alzheimer’s Disease (NCRAD).
However, variability and reproducibility is a critical challenge in human iPSC research. Biological factors, such as individual differences, genetic background, and cellular heterogeneity, contribute to this variability. Technical aspects, including variations in reprogramming efficiency, differentiation protocols, and culture conditions lead to reproducibility issues. This variability poses a substantial challenge in the accurate interpretation of data generated from iPSC models.
To overcome these barriers, we implement: 1. derivation of high-quality iPSC lines from consistent starting material with uniform non-integrating reprogramming techniques, 2. establishment of a high-quality protocols for iPSC differentiation along with clear guidelines for maintenance and storage, 3. define experimental endpoints that represent the benchmarks that a differentiating human iPSC must achieve to qualify as a differentiated neuron and 4. define robust indicators of the desired cell type, including disease biomarkers.
Since 2009, the Schuele lab has established a repository of human skin cells and fibroblasts derived from over 100 patients and healthy controls with genetic forms of Parkinson’s disease and related neurodegenerative diseases. This invaluable collection is currently housed within the Schuele lab and owned by the Michael J. Fox Foundation committed to promoting research accessibility and facilitating the broad dissemination of these invaluable cell lines across the scientific community.
The Schuele lab is affiliated with the Stanford Alzheimer Research Center (ADRC) and is supported by NIA to derive a human iPSC bank for the Stanford ADRC cohort from patients with monogenic forms of Alzheimer disease, ApoE4 genotypes with and without Alzheimer disease to study resistance and resilience, and matched healthy controls.
All human iPSCs are shared with the NINDS Human Cell and Data Repository (NHCDR), American Type Culture Collection (ATCC) or National Cell Repository for Alzheimer’s Disease (NCRAD).
Gene therapy strategies for Parkinson's disease and related neurodegenerative disorders
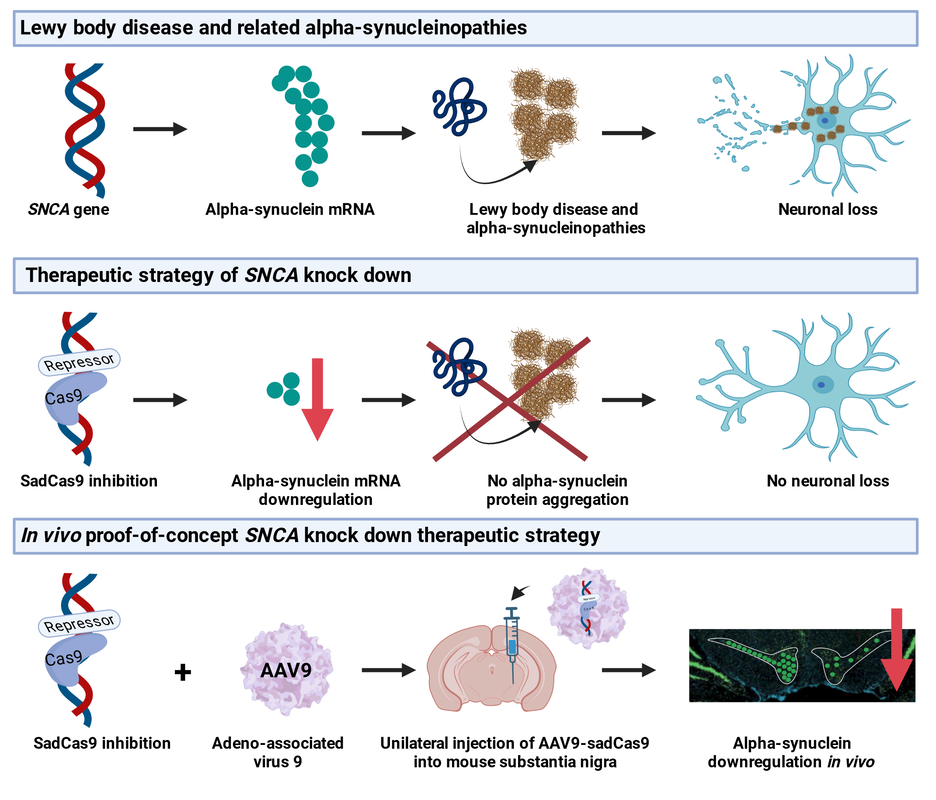
|
Gene therapy offers potentially a multifaceted approach to develop innovative and curative therapies Parkinson's disease. Firstly, it encompasses the rescue of degenerating neuronal cells by introducing genes such as glucocerebrosidase or strategies to inhibit overproduction of alpha-synuclein. Second, gene therapy approaches seek to revitalize the dopamine system by introducing genes that encode growth factors to stimulate brain cells and instigate the regrowth of the dopamine system. Lastly, gene therapies for dopamine restoration through gene-induced cell conversion, achieved by injecting a gene that orchestrates the transformation of existing cells into dopamine producing cells.
We develop gene therapies for PD and related neurodegenerative diseases that uses gene-engineering strategies. I particular we pursue a CRISPR interference approach to regulate alpha-synuclein expression levels and as collaborative project, we test an mRNA base editing strategy to convert mutant LRRk2 mRNA into wild-type sequence in vivo. We combine patient-derived iPSC models and in vivo models to test proof-of-concept and off-target effects, address efficacy and safety, and immunological responses of the virus and transgenes. |